
No-Code Fine-tuning of Chemical Foundation Models with Prithvi
Estimated Reading Time: 7 minutes

TL;DR
This week we feature a cross post from the Deep Forest Sciences blog that discusses how to use Prithvi to perform no-code fine-tuning foundation models with Prithvi.
Brief Overview of Foundation Models and LLMs
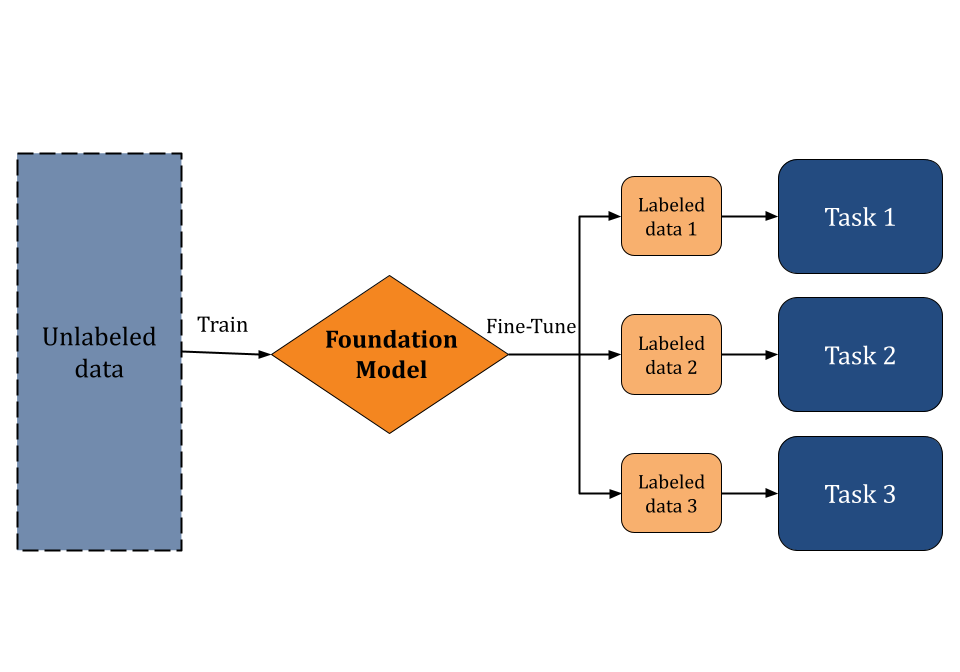
Foundation models [1] are machine learning models pretrained on large (possibly unlabeled datasets) through the use of self-supervised learning. This pre-training curriculum enables foundation models to be applied to a wide range of downstream tasks. Instead of building AI models from scratch, researchers can use foundation models as a starting point to develop specialized applications more quickly and cost-effectively. In recent years, such models have gained significant interest due to their transformative impact on fields like natural language processing, chemical analysis, and drug discovery. Foundation models that are trained on vast amounts of web data are capable of understanding and generating human-like text. Large Language Models (LLMs), such as BERT and GPT-4, are examples of popular foundation models.
Of course, foundation models don’t only have to be trained on web data. Scientific foundation models, trained on scientific data, are increasingly powerful tools for scientific discovery in general and drug discovery in particular. The SciFM initiative shows how these models can drive research and innovation by supporting collaboration across various disciplines. However such models remain difficult to use for practical scientific applications. Deep Forest Sciences’ Prithvi™ no-code platform enables scientists to fine-tune scientific foundation models for their applications without having to write sophisticated code. In this blog post, we will introduce Prithvi™’s support for no-code foundation model fine-tuning and explain how it can be a powerful tool for drug discovery.
Fine-Tuning the ChemBERTa Language Model
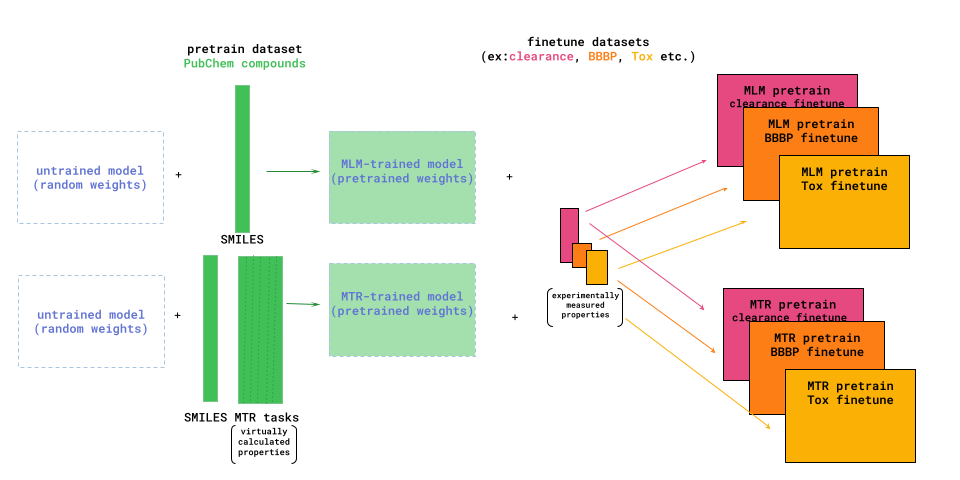
Deep Forest Sciences in partnership with the DeepChem community, has led the development of the large chemical foundation models ChemBERTa and ChemBERTa-2. ChemBERTa, based on the RoBERTa transformer model, was originally developed for molecular property prediction. This model leverages a large dataset of 77 million SMILES strings from PubChem for self-supervised pre-training. The model uses HuggingFace for streamlined pretraining and fine-tuning and is evaluated on the MoleculeNet benchmark. [2]
ChemBERTa-2 builds upon the foundations of ChemBERTa by optimizing the pretraining process [3]. The model employs masked-language modeling (MLM) and multi-task regression (MTR) to learn molecular fingerprints, leveraging the simplicity and widespread availability of SMILES strings. The pretraining improvements in ChemBERTa-2 shows that scaling pre-training datasets can significantly improve the performance of the downstream tasks.

No-Code Fine-Tuning with Prithvi
Despite their potential, fine-tuning LLMs can be complex and requires significant expertise in machine learning. Traditionally, scientists have had to rely on external engineering teams to train or fine-tune these models. To address this, Deep Forest Sciences’ has provided a no-code fine-tuning workflow on our Prithvi platform to enable biologists and chemists to train custom scientific foundation models for their needs.
A Brief Introduction to Prithvi
Prithvi™ is our AI-powered platform to accelerate small molecule drug discovery with scientific foundation models. Prithvi™ has the ability to
Perform structural analysis of targets and identify potential binding sites for a more targeted design process.
Build models based on early assay or patent data for use in large-scale virtual screens.
Construct active learning pipelines to identify and confirm high-quality hits.
Guide retrosynthetic analysis.
Suggest modifications to increase the potency, selectivity, and safety of hits.
To learn more about Prithvi™, check out our earlier blog post. In this blog post, we will focus on using Prithvi™ to fine-tune scientific foundation models.
No-Code Fine-Tuning
Prithvi offers a no-code solution for fine-tuning scientific foundation models, making advanced machine learning capabilities accessible to biologists and chemists without requiring extensive technical skills. Users can select a suitable pre-trained model from the model library and input their datasets for fine-tuning. In the rest of this tutorial, we will focus on fine-tuning a ChemBERTA-2 model with Prithvi.
Fine-Tuning ChemBERTa-2 with Prithvi
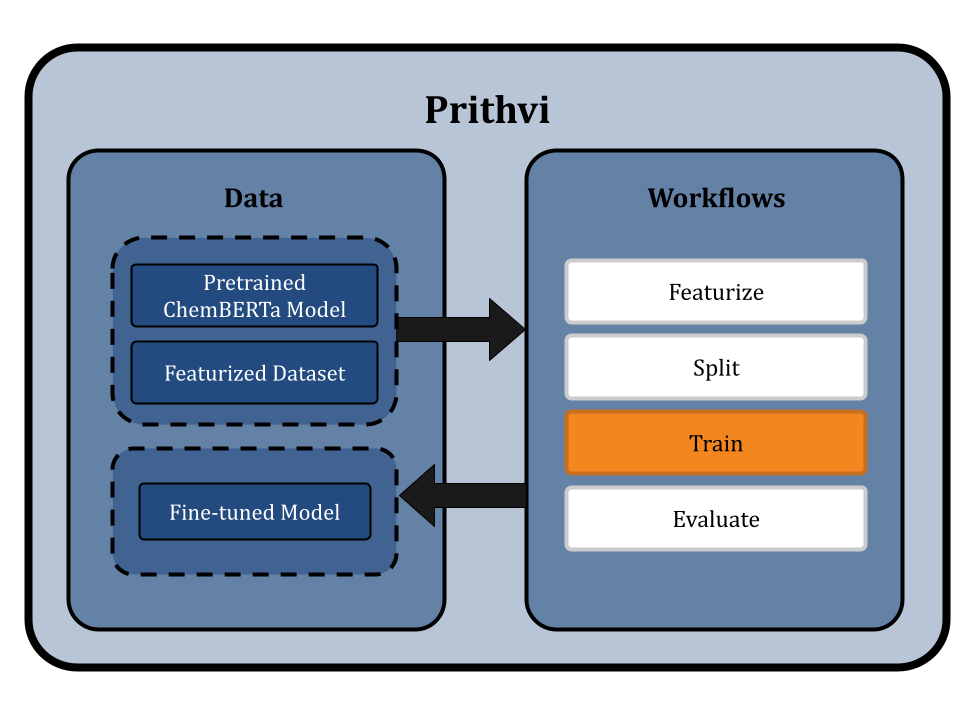
We start by uploading the dataset to Prithvi and then perform the following analysis steps:
Featurize the dataset
Splitting the dataset using scaffold split
Fine-tuning a ChemBERTa-2 Model
Evaluating the fine-tuned ChemBERTa-2
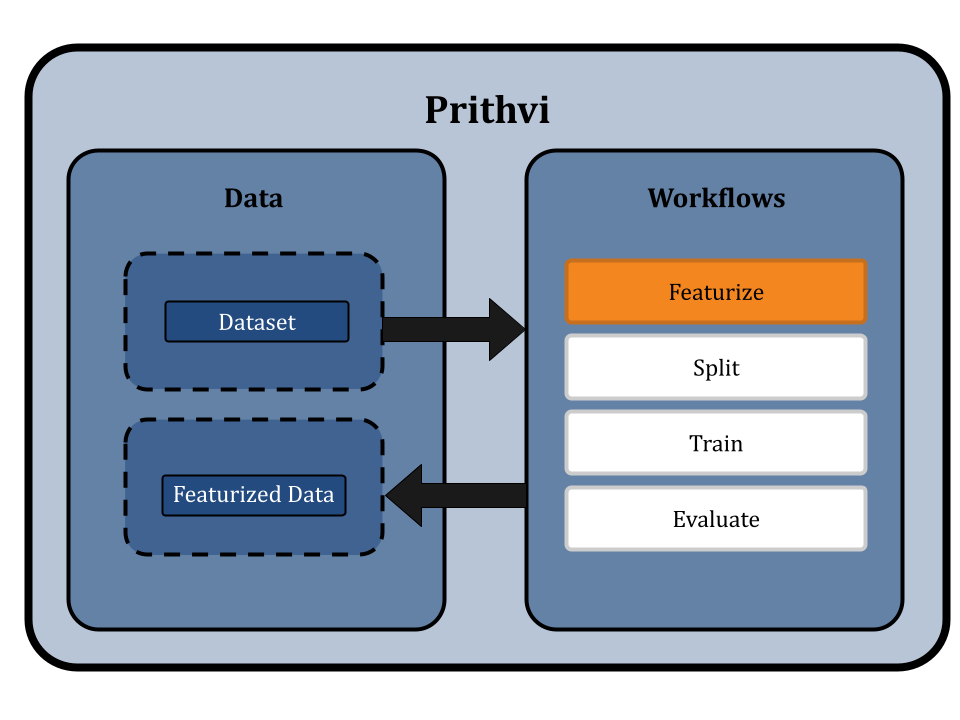
Featurize the Dataset
Featurization transforms raw data into a format suitable for machine learning. This typically involves converting data into SMILES format. For specific models like ChemBERTa-2, which directly use SMILES strings as input, traditional featurization is bypassed. We use the Dummy featurizer to featurize data for the ChemBERTa-2 model, which allows the raw dataset to be used directly without any transformation [4]. This approach preserves the detailed structural information of the molecules and simplifies the workflow to ensure that the ChemBERTa-2 model receives the most informative data for fine-tuning.
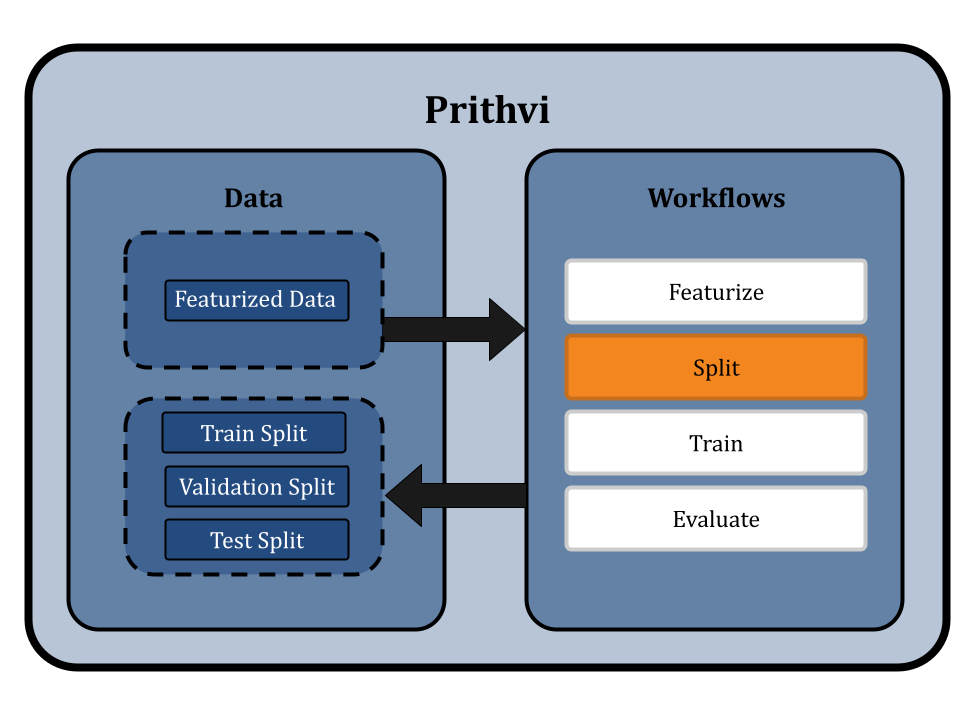
Splitting the Dataset using Scaffold Splits
Prithvi’s Split primitive is used to split datasets into train, valid, and test splits. The primitive supports different splitting algorithms like random splitting and scaffold splitting. In this workflow we use scaffold splitting to split the featurized data into train and test sets such that the molecular scaffolds in each group are as distant from the other as possible. The result is that every single molecule in the test set has a scaffold unique from the train set, and is molecularly distant from the train set. By doing this, we ensure that the test set is maximally distant from the training set, which helps to evaluate the model's generalizability and performance on new molecules proposed in a drug discovery campaign.
The default split ratio is 80:10:10. We get train, valid, and test dataset splits as output from this workflow.
Fine-tuning a ChemBERTa-2 Model
The Train primitive in Prithvi can be used to fine-tune a foundation model. Prithvi uses a default Byte-Pair Encoding (BPE) tokenizer, which breaks down large vocabularies in natural language into smaller subwords. The ChemBERTa-2 pre-training process follows the RoBERTa method, which masks 15% of tokens in each SMILES string and sets a maximum sequence length of 256 characters. This helps the model learn to predict masked tokens consisting of atoms and functional groups. The model learns the relevant molecular context for transferable tasks, such as property prediction.
The model leverages HuggingFace libraries under the hood for streamlined pretraining and fine-tuning and is evaluated on the MoleculeNet benchmark. ChemBERTa scales well with pre-training dataset size, offering competitive downstream performance on MoleculeNet and useful attention-based visualization modalities [2].
Prithvi uses a bidirectional training context with ChemBERTa-2, which has 12 attention heads and 6 layers, resulting in 72 distinct attention mechanisms. This configuration helps the model understand chemical syntax and molecular structures.
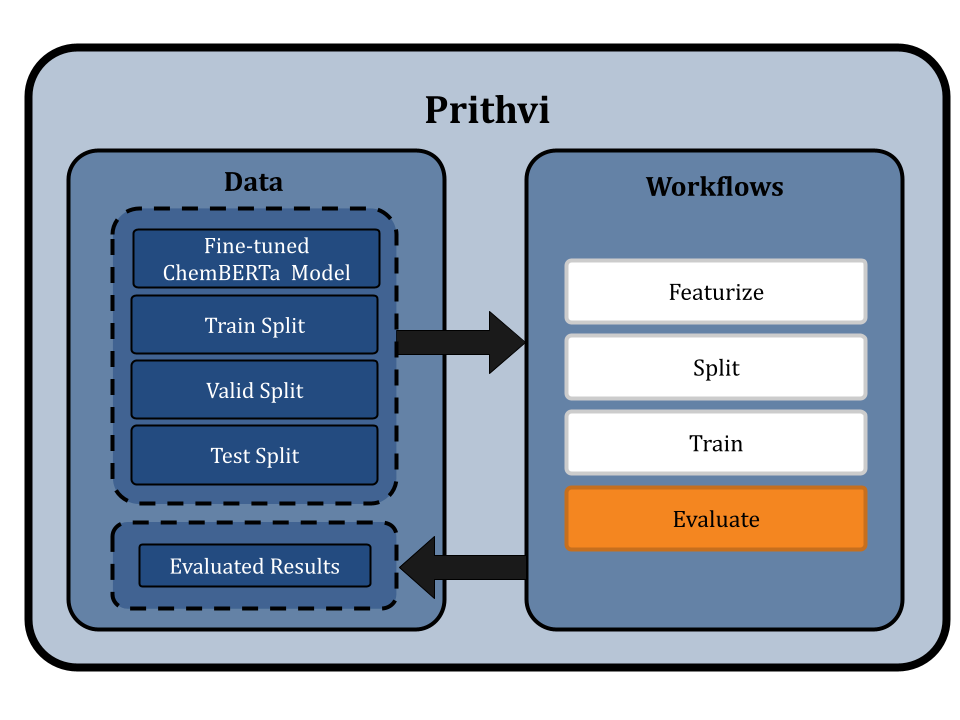
Evaluating the fine-tuned ChemBERTa-2
Prithvi’s Evaluate primitive is used to assess the performance of trained models using metrics such as the Root Mean Square Error (RMSE), which measures the differences between predicted and observed values. This evaluation can be conducted on the train, validation, and test splits as desired.
In the ChemBERTa-2 fine-tuning, we evaluate the models using the RMSE metric on the training, validation, and test splits. Multiple workflow jobs are submitted, one for each model, to ensure thorough evaluation. The evaluated results are then stored in JSON files, providing a comprehensive overview of the model's performance.
Case Study: Fine-Tuning ChemBERTa-2 on a MoleculeNet Dataset
As a case study, we use Prithvi™ to fine-tune a ChemBERTa-2 model that was pre-trained on a 10M ZINC dataset, using the Delaney dataset. The Delaney dataset, also known as ESOL (Estimated SOLubility), is a regression dataset included in the MoleculeNet benchmark. The dataset contains structures and aqueous solubility data for 1128 compounds, making it a useful benchmark for learning to predict aqueous solubility directly from a compound's structure.
After fine-tuning, we compared the results with established experimental data to evaluate the model’s performance. The MoleculeNet paper reported an RMSE score of 1.7406 ± 0.0261 (mol/L) for Random Forest (RF) Regression models and 0.8851 ± 0.0292 (mol/L) for Graph Convolutional Networks (GCN) on the Delaney dataset, using an 80:10:10 scaffold split [5]. These benchmarks were obtained through grid search-based hyperparameter tuning, a process that can be easily replicated through Prithvi.
We began by featurizing the dataset on Prithvi™ and then performed scaffold splitting through Prithvi's no-code UI.
The ChemBERTa model supports both masked language modeling (MLM) and multi-task regression for fine-tuning. In this case, we opted for a model pre-trained using MLM on the 10M ZINC dataset. The model employed a tokenizer to convert SMILES strings into input tokens, using a byte-pair encoding tokenizer trained on the PubChem10M dataset, sourced from the HuggingFace model hub.
We evaluated the fine-tuned model on train, validation and test splits using the RMSE metric. In the figure below, we compare these RMSE scores with those of the Random Forest model and the GCN model reported in the MoleculeNet paper.
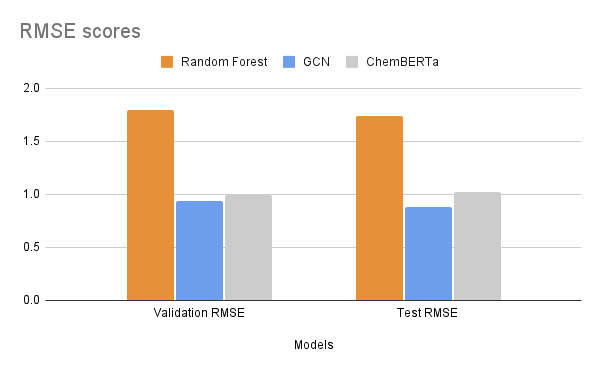
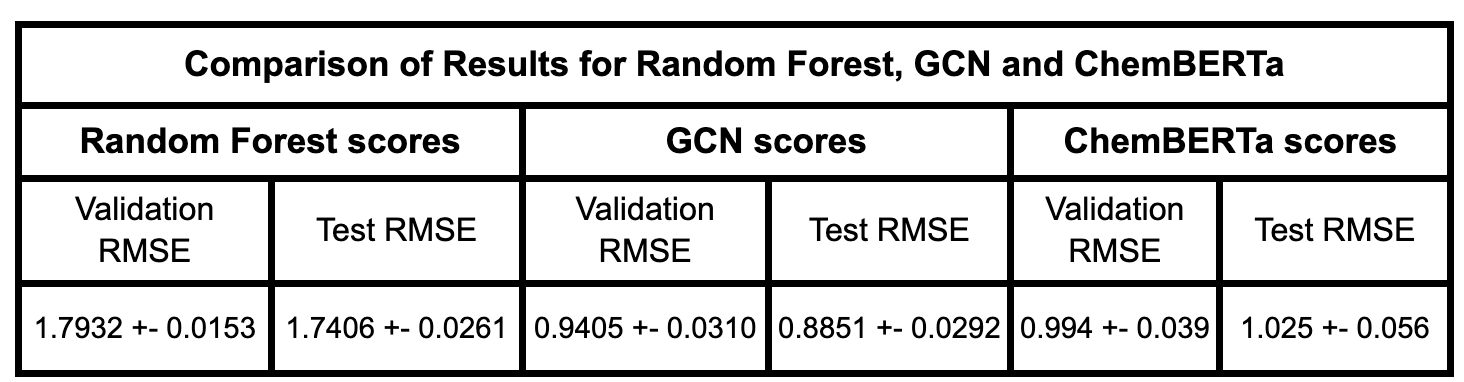
Results are comparable to those achieved with graph convolutional networks even with a small foundation model pre-trained on only 10M compound structures. The Deep Forest Sciences team is rolling out new versions of ChemBERTa, pre-trained on much larger datasets, which will enable our partners to achieve state of the art results without having to write any code.
Conclusion and Takeaways
Deep Forest Sciences' Prithvi™ platform offers a no-code solution for fine-tuning scientific foundation models like ChemBERTa for different downstream molecular property prediction tasks. It provides a user-friendly interface and pre-trained models to enable researchers to train their own custom scientific foundation model and simplifies the complex process of model fine-tuning, making these methods accessible to biologists and chemists without extensive machine learning expertise. Prithvi™ will help scientists design and discover powerful new drugs by making cutting edge AI tools accessible and friendly.
References
[1] Bommasani, R., et al. (2021). On the opportunities and risks of foundation models. Center for Research on Foundation Models (CRFM), Stanford University. https://crfm.stanford.edu/assets/report.pdf
[2] Chithrananda, S., Grand, G., & Ramsundar, B. (2020). "ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction." https://arxiv.org/abs/2010.09885
[3] Chithrananda, S., Grand, G., & Ramsundar, B. (2022). “ChemBERTa-2: Towards chemical foundation models.” https://arxiv.org/abs/2209.01712
[4] ”Featurizers”. DeepChem Documentation. https://deepchem.readthedocs.io/en/latest/api_reference/featurizers.html#dummyfeaturizer
[5] Wu, Z., Ramsundar, B., Feinberg, E. N., et al. (2018). MoleculeNet: A benchmark for molecular machine learning. arXiv. https://arxiv.org/abs/1703.00564
About Deep Forest Sciences
Deep Forest Sciences’ no-code AI Prithvi™ toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational no-code AI technology to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com to learn more!
Feedback and Comments
Please feel free to email me directly (bharath@deepforestsci.com) with your feedback and comments!
About
Deep Into the Forest is a newsletter by Deep Forest Sciences, Inc. We’re a deep tech R&D company building Chiron, an AI-powered scientific discovery engine for the biotech/pharma industries. Deep Forest Sciences leads the development of the open source DeepChem ecosystem. Partner with us to apply our foundational AI technologies to hard real-world problems in drug discovery. Get in touch with us at partnerships@deepforestsci.com!
Credits
Author: Bharath Ramsundar, Ph.D.
Editor: Sandya Subramanian, Ph.D.
Cross-Post Research and Writing: Riya Singh
Research and Writing: Rida Irfan
Great article. Do you think an MoE approach could foster an open(ish) ecosystem in which researchers in academia/industry can contribute and share individual models? Downstream aggregators could pick and choose from this marketplace based on their specific requirements (drug discovery, diagnostics, predicting protein interactions, etc.). Lots of legal, ethics, financial issues to work out in addition to technical ones.